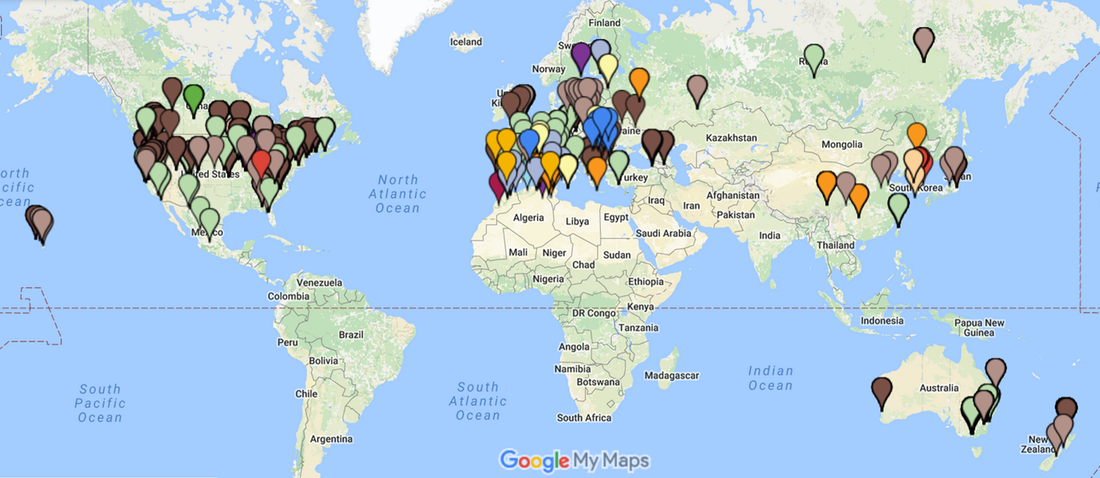
So far we have received >2,000 butterflies from 30 US states and 32 different countries! The project is still ongoing, so if you catch a few, send them in!
We've now sequenced part of the genome of >600 of the butterflies sent in
|
|
|
Turning your samples into genetic data =)

Crushing up the legs to allow our extraction buffer to break apart (lyse) the cells and release the DNA inside.
|
First, we started by extracting DNA from the leg tissue of each butterfly. This was done by crushing up the legs and putting them in what is called an "extraction buffer" - basically detergent (soap) and salt solution. The detergent helps lyse (break apart) the cells and release their DNA into the solution. We also added an enzyme (protein) called Proteinase K that digests all the proteins that are also inside the cells that might otherwise degrade the DNA. We then let this incubate overnight at 56° C. This helps denature (unfold) the proteins and makes Proteinase K's job easier...chomp, chomp, chomp. We don't want proteins, they will just get in the way and ruin (degrade) our DNA. Eat Pro K, eat away. =)
Making the "extraction buffer"
|
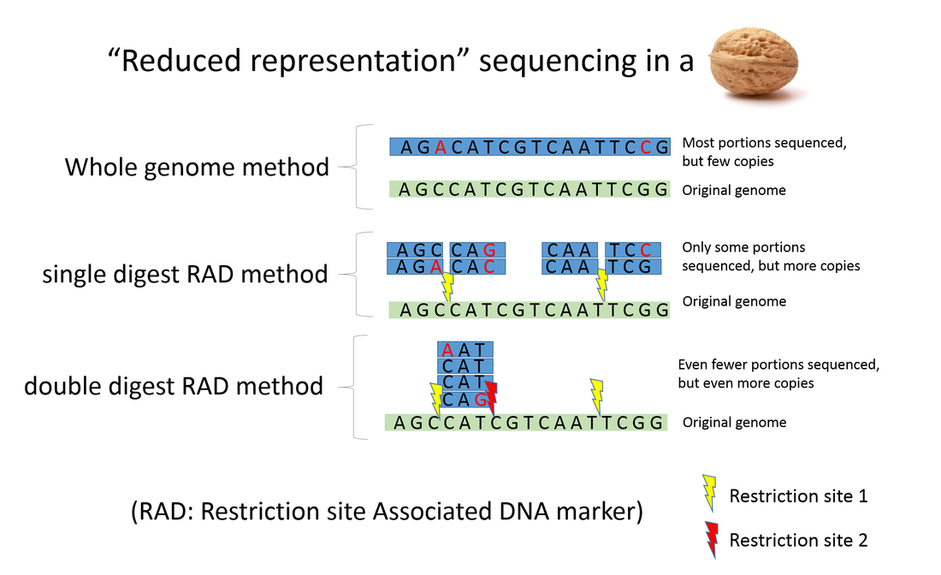
We used what is called a "double-digest RAD" method to prepare our DNA for sequencing. In a nutshell this method allows us to sequence a subset of the genome instead of the whole thing. Why sequence a subset and not the whole genome? Well, many of the sequencing machines only sequence a set # of base pairs (A's, C's, G's, and T's). The Pieris rapae genome is made of about ~240 million base pairs! So if the machine produced 350 million base pairs, we might have a little more than one copy ("1X coverage") of the whole genome. Sounds good right? Unfortunately, these machines make mistakes, therefore we need multiple copies so we can tell what is a mistake (for example if we have 5 copies of a part of the genome, with 4 that read AGGTC at a given position, and 1 that reads AGGTA, we can identify the last bp as a mistake). How do we make sure we get multiple copies? Instead of sequencing very few copies of entire genome, we can sequence fewer portions of the genome, but get more copies of each portion. How do we do this?
There are proteins called "restriction enzymes" that can cut specific sequences called a restriction sites (for example the enzyme EcoR1 cuts every time it finds the sequence GAATTC in the genome), and these restriction sites are scattered throughout the genome. Using these enzymes we direct our DNA to only sequence regions of the genome near these restriction sites. The double-digest method uses two restriction enzymes, only sequencing regions where two different restriction sites are found, and therefore sequences even less of the genome, but produce even more copies. Why would we want to sequence even less of the genome? Well in our case we want to sequence multiple (250) different butterflies, so we have lots of genomes to sequence and therefore the sequencing has to be shared - the copies will have to be divided by the # of butterflies. To ensure we get enough copies for every butterfly, we sequence less of the genome. In the end this still allows us to sequence thousands of different parts of the genome, which is still plenty to answer lots of interesting evolutionary questions =)
There are proteins called "restriction enzymes" that can cut specific sequences called a restriction sites (for example the enzyme EcoR1 cuts every time it finds the sequence GAATTC in the genome), and these restriction sites are scattered throughout the genome. Using these enzymes we direct our DNA to only sequence regions of the genome near these restriction sites. The double-digest method uses two restriction enzymes, only sequencing regions where two different restriction sites are found, and therefore sequences even less of the genome, but produce even more copies. Why would we want to sequence even less of the genome? Well in our case we want to sequence multiple (250) different butterflies, so we have lots of genomes to sequence and therefore the sequencing has to be shared - the copies will have to be divided by the # of butterflies. To ensure we get enough copies for every butterfly, we sequence less of the genome. In the end this still allows us to sequence thousands of different parts of the genome, which is still plenty to answer lots of interesting evolutionary questions =)
